大伙儿可能都听说过PCR,尤其是这几年,它在各种检测中可真是出了名。但说实话,很多人对它的了解大概就停留在“一种能复制DNA的神奇技术”这个层面。今儿个咱们就掰开揉碎了唠唠,这PCR扩增技术的过程到底是咋回事,为啥它这么重要,以及在实验室里摆弄它的时候,又有哪些门道和容易踩的坑。
PCR到底是个啥?为啥离了它不行?
简单来讲,PCR,中文叫聚合酶链式反应,它的能耐就是在试管里,短短几小时内,把一段我们感兴趣的DNA片段像变魔术似的复制出成千上万甚至上亿份-6。你可以把它想象成一个专一且高效的DNA复印机。
这项技术发明于上世纪80年代,它的发明者还因此拿了诺贝尔奖,可见其有多厉害-10。现在,从医学诊断(比如某些病毒检测)、基因研究,到法医鉴定、食品安全检查,甚至咱们做亲子鉴定,到处都有它的身影-9-10。可以说,没有PCR,现代分子生物学和很多相关领域的研究,基本就玩不转了。
核心三步走:变温舞蹈的精髓
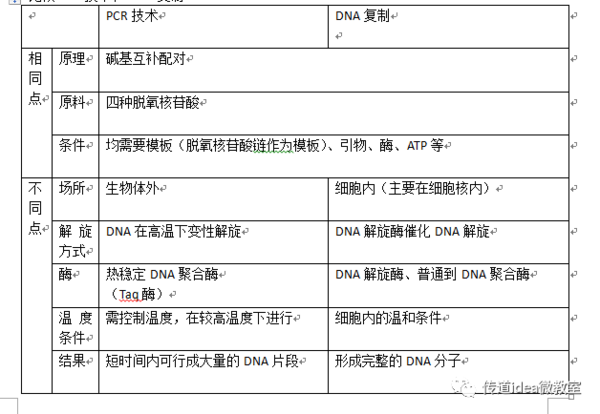
这PCR扩增技术的过程,本质上就是一场精心编排的“变温舞蹈”,围绕着三个核心步骤循环往复:变性、退火、延伸。这三个词听起来专业,其实道理挺简单-2-8。
第一步,高温变性——把双链拆开。 这就像把一根双股拧成的绳子(DNA双螺旋)放在开水里烫一下,绳子受热就松开了,变成了两条独立的单链-4。在PCR仪里,这个温度通常设定在94°C左右,维持几十秒,目的就是让作为模板的DNA双链彻底分开,暴露出内部的碱基序列,为下一步做准备-5。
第二步,低温退火——让引物精准定位。 温度降下来,通常降到55°C左右(这个温度很关键,后面会细说)。这时候,反应体系里预先加进去的两种“引物”就开始发挥作用了。引物是人工合成的一小段DNA单链,它的序列设计得和咱们想要复制的目标DNA片段的两头,是完全互补匹配的-2。温度一合适,这两个小引物就像特制的“钩子”或者“向导”一样,分别精准地钩到两条模板单链的对应位置上,准备“引路”-8。这一步决定了PCR的特异性——复制得准不准,全看引物和模板配不配。
第三步,中温延伸——合成新链条。 温度再升到72°C左右,这是体系里用的Taq DNA聚合酶(一种耐热的“复制工”)干活最舒服的温度。酶会以单链为模板,从引物“钩住”的地方开始,按照A配T、C配G的配对规则,把体系里游离的原料(四种脱氧核苷酸,dNTP)一个个接上去,合成一条全新的、与模板链互补的DNA链-2-4。
就这样,经过“加热分开 - 冷却结合 - 温合延伸”一个循环,一段目标DNA就变成了两段。 接下来,机器会自动进入下一个循环,新生成的两段DNA又会被当作模板,再次被复制。如此反复,像链式反应一样,经过25到35个循环后,目标DNA的数量就能从最初的几个拷贝,呈指数级增长到数百万甚至数亿个拷贝,足够我们进行各种分析了-5-9。
一锅好汤:反应体系里的“配料”学问
光有程序还不行,PCR这锅“汤”能不能煲好,里面的“配料”——也就是反应体系——至关重要。这可是PCR扩增技术的过程能够顺利启动和进行的物质基础,配不好,后面跳啥舞都白搭。
模板DNA: 就是含有你要复制的那段“目标”的DNA。它的纯度不能太差,不能混有抑制聚合酶活性的东西,但用量却极少,有时甚至一个细胞的DNA都够用,可见PCR有多灵敏-9-10。
引物: 这是决定实验成败的“灵魂配料”。一对好的引物,长度一般在20个碱基左右,序列要特异,不能自己和自己或者互相之间乱结合(形成二聚体),而且两条引物的“退火温度”要差不多-8。设计引物是个技术活,现在大多靠专业软件。
Taq DNA聚合酶: 从耐热细菌里来的“劳模”。普通的DNA聚合酶一加热就失活了,而它能在PCR反复的高温中保持活性,是实现自动循环的关键-10。
dNTP: 就是合成新链的原料,腺嘌呤、鸟嘌呤、胞嘧啶、胸腺嘧啶这四种脱氧核苷酸,缺一不可,而且浓度要均衡-8。
镁离子(Mg²⁺): 别看它不起眼,却是聚合酶干活必需的“助工”。它的浓度高低会直接影响酶的活性和复制的精确度,通常需要反复摸索找到最佳浓度-8。
缓冲液: 提供一个稳定的酸碱度和离子环境,让各种“配料”能和谐共处,高效工作-9。
把这些东西按比例小心翼翼地在冰上混合好(防止酶提前活动),再加入一滴矿物油或使用有热盖的PCR仪防止液体蒸发,就可以上机运行程序了-1-5。
温度与时间:实验室里的“手感”微调
了解了基本流程和配方,你会发现,在真实的实验室操作中,PCR扩增技术的过程充满了需要根据实际情况微调的“艺术”。很多时候,照着标准配方做不出来,问题就出在这些参数的细节上。
退火温度(Tm值): 这是最常需要优化的参数。前面说的55°C只是一个常见起始值。如果退火温度太高,引物钩不上模板;温度太低,引物就可能“瞎钩”,产生一堆没用的非特异性产物。通常,退火温度要比引物的理论“解链温度”(Tm值)低个3-5°C-8。如果一次要优化多对引物(比如做多重PCR),或者不确定最佳温度,可以用PCR仪的“梯度PCR”功能,在同一块板子上设置一排不同的退火温度同时跑,哪个孔的结果好就用哪个温度,省时省力-3。
循环次数: 不是越多越好。一般25-35个循环足够了-8。循环太多,后期原料不足,酶也累了,容易产生错误,而且会进入“平台期”,产量不再增加-8。
延伸时间: 这取决于你要复制的DNA片段有多长。通常按照Taq酶每分钟合成1000-2000个碱基的速度来估算-8。复制长片段,时间就要给足。
优化这些条件,往往是个试错的过程。实验室新手常会对着电泳凝胶上模糊的一片(非特异性条带)或者空空如也(没扩增出来)的结果发愁,这时候就得回头检查引物设计、调整Mg²⁺浓度或者优化退火温度了。
终点与验证:看见结果才算成功
程序跑完,机器“嘀”的一声响,并不意味着工作结束。我们怎么知道扩增成功了,而且扩增出来的就是我们要的东西呢?这就需要对产物进行检测和分析。
最经典、最常用的方法就是琼脂糖凝胶电泳-1-5。简单说,就是让PCR产物在通电的凝胶里“跑步”,分子量小的跑得快,大的跑得慢。同时点上已知长度的标准品(DNA Marker)做对照。跑完胶后,用一种叫溴化乙锭的染料(在紫外灯下会发光)染色,放到紫外凝胶成像仪下一照。如果在我们预期大小的位置出现了一条清晰、明亮的条带,而阴性对照(不加模板)没有,那基本就说明PCR成功了-5-9。
如果条带位置不对、有多条带或者有拖尾,那就可能意味着有非特异性扩增、引物二聚体或者模板降解等问题,又得回到优化步骤了。
对于要求更高的实验,比如后续要进行基因克隆或者测序,可能还需要对PCR产物进行纯化,去除残留的引物、酶和dNTP等,甚至要用特定的酶给产物和载体DNA切出匹配的末端,再进行连接-1。通过测序来百分百确认扩增序列的准确性,因为即便是高效的Taq酶,在复制过程中也有极低的出错概率-9。
所以说,PCR扩增技术的过程,远不止是设置好机器按下开始键那么简单。它是一套从原理理解、精心设计、精确配液、参数优化到结果验证的完整逻辑和实践体系。每一次成功的扩增背后,可能都藏着实验员对细节的无数次打磨。希望这篇唠嗑,能帮你把这套流程理得更清楚些,无论是为了学习、应用,还是纯粹满足好奇心。